Flow Cytometry Core Facility - Best Practices
The principles of a sort
The Fusion sorts by aspirating cells from the sample tube to combine them with the stream of sheath fluid (commonly FACSFlow). At the flow cell, the stream passes the interrogation point where lasers interact with the cells. The instrument electronics calculates the location for each cell and determines whether it meets the sort criteria. A transducer in the instrument induces droplets to form in the stream using vibration. Cells in the stream are incorporated within the droplets. If a cell meeting the sort criteria is in the final drop before the break off, the instrument will charge that droplet and this will be deflected into the collection container by the charge plates.
Cell sorting is a highly complex process with many variables effecting sort time, yield, purity and viability. Because cells may respond poorly to the sorting process, so optimisation of the sorting conditions for best survivability may be a critical and time consuming first step.
Biosafety
All users initiating sorts with new cell lines or primary cells must provide evidence of completed GMO forms. The Imaging Facility is serious about biosafety.
The Fusion is housed a bio-safety cabinet. Sterilisation of the Fusion can be completed by running a preparation for aseptic sort protocol upon request. To minimize microbial contamination of your sort, as a basic practice phosphate buffered saline (FACSFlow includes sodium azide) is filtered through a 0.2 µm filter. At the end of a biohazardous sort we clean the sample lines with FACSClean (bleach), FACSRinse (detergent) and as part of the shutdown procedure 70% Ethanol is rinsed through all fluidic lines. We can run 70% ethanol between sorts to prevent cross-contamination.
Sample Preparation
Notoriously the most important step in any flow cytometry experiment. Fluorescently activated cell sorting works best when cells are in a single cell suspension that is easily maintained during the entire sort. Clumps, debris and doublets will lower the efficiency and purity of a sort and may clog the instrument. Significant time can be wasted if the instrument clogs due to cleaning steps and recalibrating the drop delay profile. Nozzle clogs can be so severe that rectifying the instrument requires an engineer call out. Strain cells that are prone to clumping through a 35µm strainer before sorting.
To stain your cells the recommended staining buffer is 1x Phosphate Buffered Saline (PBS) or Hank’s Balanced Salt solution (HBSS) with 1-5% BSA or FCS.
To dissociate adherent cells from culture flasks, EDTA should be at a final concentration 5 mM. OAdherent cells are typically dissociated using trypsin or some type of EDTA treatment. Adherent cells can be a problem when sortedooften adherent cells are trypsinised, then inactivated by adding fetal calf serum that reforms divalent cations but his encourages cells to clump. Rather, consider using a trypsin inhibitor instead of serum. Cells can be killed during trypsinisation process, so DNAse I (100 µg/ml, or 10 units/ml) or II can be added to prevent free DNA sticking to cells causing them to aggregate. Accutase is an alternative solution that gently detaches confluent cells from plasticware and does not have to be neutralized like trypsin and epitopes that can be stained for flow cytometry are preserved. To prevent cells from clumping, centrifuge cells between 300g to 800g for 1-3 minutes in a round bottom 96 well plate or tube.
On all sorts, we run an analysis template that applies a double doublet discrimination method to ensure multiples of cells passing through the laser intercept simultaneously are excluded. In addition the Fusion has an agitation function in the sample chamber, this can be used to prevent cells settling out and clumping.
Please bring your cells in PBS or FACSFlow buffer. Media containing phenol red will increase background fluorescence. Most auto-fluorescence is generated by the 488 nm (blue) laser line. If you suspect that your cells will have high background fluorescence, we recommend that you use red-excitable (633 nm laser line) fluorochromes such as APC or APC-Cy7 because cells tend to demonstrate less auto-fluorescence when emission is measured past 650nm. You know the best optimal conditions for keeping your cells healthy and alive. If you expect your cells will do better in their culture media then do bring your cells in media. Sorting dead or unhealthy cells is a waste of time, reagents and money.
The Fusion has a temperature control unit, we can set temperatures for the sample chamber as well as the collection chamber. If your cells are particularly sensitive, request a set temperature is used.
The best results will be obtained using just enough protein to keep your cells happy but at low enough concentrations to not significantly affect light scattering. High protein concentrations in sample buffer may create a refractive bias because of the increased optical density and refractive index of the buffer containing protein. Laser light passing through buffers having different refractive indices may cause distortion of the light scatter signals.
Cell Concentrations
To sort cells as quickly and efficiently as possible samples should not be too dilute or concentrated, these conditions may adversely affect the yield and purity of your sort. The sorter keeps track of where your cells are as they pass through the laser intercepts and get incorporated into the last attached droplet. Cells in a high concentration will be in close proximity and the instrument may struggle to identify droplets appropriate to sort, or deem them too close to a droplet that would have been sorted and so rejects them to save contaminating purity. This is called a coincidence abort. We can adjust the sorting criteria towards purity by sacrificing yield, or vice versa.
If your cell preparation is too dilute, we may have to speed up the sample acquisition rate. This widens the core stream which increases the coefficient of variation (CVs) and lowers the fluorescence sensitivity of the instrument. Lower sensitivity is detrimental to the quality, especially if your cells are not brightly stained for your sort marker.
Recommended cell concentrations
| Nozzle size | Cell Type | Concentration |
|---|---|---|
| 70 μm | Lymphocytes, small cells | 7.5 - 12 x 106 cells/ml |
| 85 μm | <15 μm in diameter cell lines | 5 - 7.5 x 106 cells/ml |
| 100 μm | <15 μm in diameter cell lines | 1 - 7.5 x 106 cells/ml |
If your cells tend to aggregate, you may want to further lower your concentration to prevent clumping.
When sorting single cells into a plate, the Fusion has a sort index function that means you can trace which cell on your plot ended up in each specific well. This traceability function can be very useful.
Controls
Please bring the proper experimental controls, for example unstained cells, single fluorescently stained cells or beads, fluorescence minus one, negative controls. We cannot gate properly without adequate controls. For multi-color experiments you must bring compensation controls or design your experiment with fluorochromes that do not spectrally overlap.
Labelling your cells or particle of interest with fluorochromes and dyes
Several Spectral Viewers are available online to help design your panel of dyes, and understand where the emission spectrum will lay in relation to the laser filters that we have available.
These include:
Sorters at the RVC
The RVC Imaging Facility provides access to a FACS AriaFusion™. This equipment contains four lasers combined with a flow cell that gives rise to excellent fluorescence resolution.
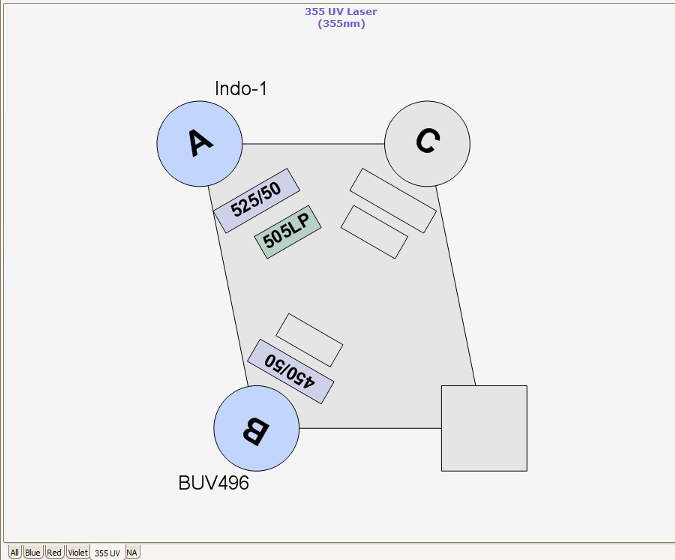
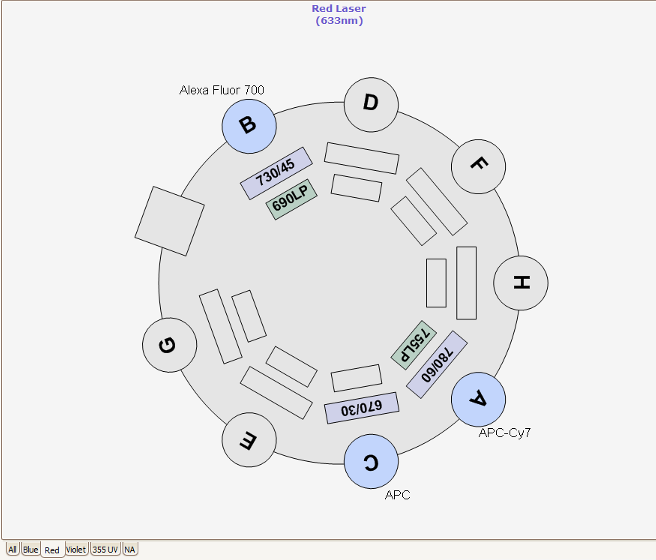
Nozzles, Sort Pressure, Cell Size and Morphology
Three different nozzle sizes (70, 85 and 100 um) are available on the RVC Fusion. Best practice is to select the nozzle 5x the diameter of the cell to be sorted. For each nozzle the optimal sheath pressure is: 70 μm = 70 PSI, 85 μm = 45 PSI and 100 μm = 20 PSI. The RVC do not currently have 130nm nozzles, however 10-11 PSI sorting can be problematic because this is towards the sorter pressure regulator’s lower boundary and slower stream velocities can give rise to random and uneven droplet formation and sorts much slower. Lymphocytes are viable after being sorted at 70 PSI, unfortunately many primary cells and certain cell lines are significantly less viable after high speed, high pressure sorting. For these cell lines, we recommend sorting with the 100 μm nozzle at 20 PSI.
Particles of all shapes and sizes of cells can be sorted. Small, spheroid cells are easily incorporate into droplet, so they sort best. Large, irregular or elongated cells are much more difficult to incorporate into droplets, so they sort less efficiently. We carefully calculate the droplet break-off point to ensure cells do not miss the collection tube, contaminating other collection tubes and negatively impact sample purity.
Excluding Dead Cells
When cells are dead or dying, they flip their membrane and so appear to express markers that were previously not a cell surface level. Check your cell viability before arriving at the FACS. LIVE/DEAD stains can be used, Propidium iodide (0.5 – 2.0 µg/ml), 7-AAD (ex 488, em 647), Annexin V (ex 535, em 617) or DAPI (ex 405, em 461) all of which are viability reagents and will allow us to gate out dead cells. However, PI can be useful because it is so bright it does overlap with PE, Texas Red, PerCP,Cy3 etc.
Sorting Dim Populations
Sorting works best when the population to be sorted is much brighter than its negative control. Tight gates should be set on dim cells. Dimly stained cells have fewer attached fluorochromes for the lasers to excite, fewer photons gathered by the light collecting optics and fewer photons generated by the photo-multiplier tubes. Dim cells can have larger standard deviations, so a cell that appears positive in the sort window may appear negative in a post-sort analysis. It is best to bias these sorts toward purity over yield.
If you suspect that your marker of interest is present in low levels on your cells, use the brightest fluorochromes like Brilliant Violets, UV fluorochromes or PE and APC. You may also try staining your primary antibody with a labeled secondary antibody to amplify fluorescence.
Cell Recovery after Sort
For best result sort into cell culture media containing higher than normal concentrations of serum or appropriate protein. Sorted cells are in droplets and this PBS will dilute out the protein in your collection media. Adding 10 mM HEPES buffer to the media can buffer the CO2 in the collection media. Adding antibiotics to recovery media will cut down on the risk of microbial contamination.
The temperature control unit can be used to warm the collection tubes or culture plates.
Polypropylene FACS tubes encourage better recovery because they have less inherent charge. Polystyrene can hold a charge and can deflect charged droplets containing your sorted cells, but to improve recovery pre-coat your collection tubes with media, serum, 1% BSA or buffer containing protein. Because the droplet incorporating the sorted cell is charged, the droplet may become attracted to the charge of the plastic, stick to the side, small volumes of liquid evaporate and cells die. Pre-coating collection tubes with protein eliminates the charge on the plastic collection tubes.
When sorting a rare population, it may be faster to sort twice, once for enrichment and secondly, for purity.
Post-sort Evaluation
If the final volume of sorted cells permits, the collection tube will be reloaded into the sample chamber to evaluate the yield and purity of the sorted cells. This confirms that we have collected the intended cells of interest. If port-sort death is an issue, a LIVE/DEAD stain could be added to a small volume of sorted sample to assess viability. You may request this is not performed if your sorted sample is precious.