Reconstruction of ancient chromosomes offers insight into mammalian evolution
What if researchers could go back in time 105 million years and accurately sequence the chromosomes of the first placental mammal? What would it reveal about evolution and modern mammals, including humans?
Researchers from the RVC in the UK collaborated with academics from University of California, Davis (UC Davis), Carnegie Mellon University in Pittsburgh, Konkuk University in Seoul and the University of Illinois at Urbana–Champaign, to go back in time by computationally recreating the chromosomes of the first eutherian mammal, the long-extinct shrew-like ancestor of all placental mammals.
The findings are published in Proceedings of the National Academy of Sciences, and have broad implications for understanding how chromosomal rearrangements over millions of years may contribute to human diseases, such as cancer.
Harris Lewin, a lead author of the study and a professor of evolution and ecology at UC Davis, said: “The revolution in DNA sequencing has provided us with enough chromosome-scale genome assemblies to permit the computational reconstruction of the eutherian ancestor, as well as other key ancestors along the lineage leading to modern humans. We now understand the major steps of chromosomal evolution that led to the genome organization of more than half the existing orders of mammals. These studies will allow us to determine the role of chromosome rearrangements in the formation of new mammal species and how such rearrangements result in adaptive changes that are specific to the different mammalian lineages.
“By gaining a better understanding of the relationship between evolutionary breakpoints and cancer breakpoints, the essential molecular features of chromosomes that lead to their instability can be revealed. Our studies can be extended to the early detection of cancer by identifying diagnostic chromosome rearrangements in humans and other animals, and possibly novel targets for personalized therapy.”
Descrambling chromosomes

To recreate the chromosomes of these ancient relatives, the team began with the sequenced genomes of 19 existing placental mammals — all eutherian descendants — including human, goat, dog, orangutan, cattle, mouse and chimpanzee.
The researchers then used a new algorithm they developed called DESCHRAMBLER which computed (“descrambled”) the most likely order and orientation of 2,404 chromosome fragments that were common among the 19 placental mammals’ genomes.
Jian Ma, the study’s co-senior author and an associate professor of computational biology at Carnegie Mellon University, said: “It is the largest and most comprehensive such analysis performed to date, and DESCHRAMBLER was shown to produce highly accurate reconstructions using data simulation.”
In addition to the eutherian ancestor, reconstructions were made for the six other ancestral genomes on the human evolutionary tree: boreoeutherian, euarchontoglires, simian (primates), catarrhini (Old World monkeys), great apes and human-chimpanzee. The reconstructions give a detailed picture of the various chromosomal changes that have occurred over the 105 million years between the first mammal and Homo sapiens.
Rates of Evolution Vary
One discovery is that the first eutherian ancestor likely had 42 chromosomes, four less than humans. Researchers identified 162 chromosomal breakpoints – locations where a chromosome broke open, allowing for rearrangements — between the eutherian ancestor and the formation of humans as a species.
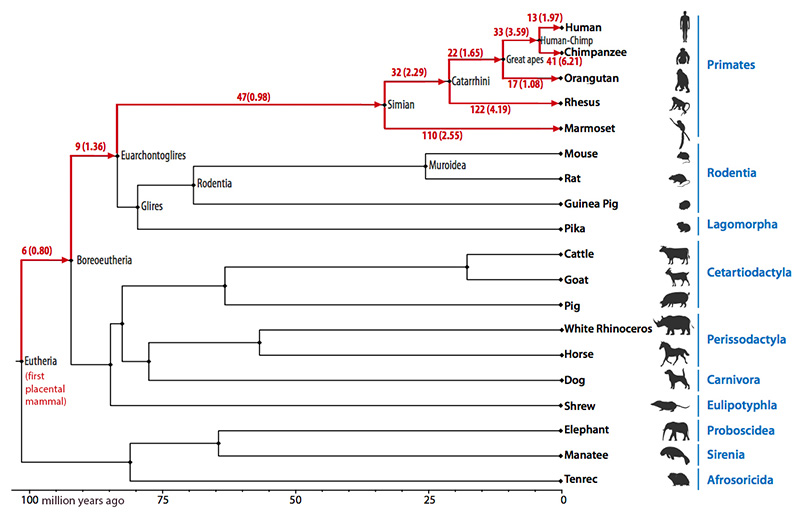
The rates of evolution of ancestral chromosomes differed greatly among the different mammal lineages, but some chromosomes remained extremely stable over time. For example, six of the reconstructed eutherian ancestral chromosomes showed no rearrangements for almost 100 million years until the appearance of the common ancestor of human and chimpanzee.
Orangutan chromosomes were found to be the slowest evolving of all primates. In contrast, the lineage leading to chimpanzees had the highest rate of chromosome rearrangements among primates.
Dr Denis Larkin, co-senior author of the study and a reader in comparative genomics at the RVC, said: “When chromosomes rearrange, new genes and regulatory elements may form that alter the regulation of expression of hundreds of genes, or more. At least some of these events may be responsible for the major phenotypic differences we observe between the mammal orders.”
The chromosomes of the oldest three ancestors (eutherian, boreoeutherian, and euarchontoglires) were each found to include more than 80 percent of the entire length of the human genome, the most detailed reconstructions reported to date. The reconstructed chromosomes of the most recent common ancestor of simians, catarrhini, great apes, and humans and chimpanzees included more than 90 percent of human genome sequence, providing a structural framework for understanding primate evolution.
Dr Marta Farré Belmonte, Postdoctoral Research Assistant at the Royal Veterinary College and co-first author on the paper, said: "It is the first time that the history of each mammalian chromosome is delineated in such great detail, demonstrating that some chromosomes were maintained in all the species, while others changed dramatically among the different eutherian lineages."
Research Reference
Jaebum Kim, Marta Farré, Loretta Auvil, Boris Capitanu, Denis M. Larkin, Jian Ma, and Harris A. Lewin. Reconstruction and evolutionary history of eutherian chromosomes PNAS 2017 ; published ahead of print June 19, 2017, doi:10.1073/pnas.1702012114
The source code and input and output files for the algorithm developed for this research can be downloaded from DESCHRAMBLER.
Press Office Contact
Uche Graves / Zoe Chadwick
T: 0800 368 9520
E: uche.graves@plmr.co.uk / zoe.chadwick@plmr.co.uk
Notes to Editors
The academics who contributed to this study are:
- Harris Lewin, University of California, Davis
- Jian Ma, Carnegie Mellon University in Pittsburgh
- Denis Larkin, RVC
- Marta Farré Belmonte, RVC
- Jaebum Kim, Department of Biomedical Science and Engineering at Konkuk University in Seoul, South Korea
- Loretta Auvil, Illinois Informatics Institute at the University of Illinois at Urbana–Champaign in Urbana, Illinois
- Boris Capitanu, Illinois Informatics Institute at the University of Illinois at Urbana–Champaign in Urbana, Illinois
The research was supported by:
- the Biotechnology and Biological Sciences Research Council
- the Robert and Rosabel Osborne Endowment (UC Davis)
- the National Institutes of Health
- the National Science Foundation
- the Ministry of Science, ICT & Future Planning of Korea
- the Ministry of Education of Korea
- the Rural Development Administration of Korea
- This work was conducted as a contribution to the Genome 10K Project.
The Royal Veterinary College (RVC) is the UK's largest and longest established independent veterinary school and is a constituent College of the University of London. The RVC offers undergraduate, postgraduate and CPD programmes in veterinary medicine, veterinary nursing and biological sciences, being ranked in the top 10 universities nationally for biosciences degrees. It is currently the only veterinary school in the world to hold full accreditation from AVMA, EAEVE, RCVS and AVBC.
A research-led institution, in the most recent Research Excellence Framework (REF2014) the RVC maintained its position as the top HEFCE funded veterinary focused research institution.
The RVC also provides animal owners and the veterinary profession with access to expert veterinary care and advice through its teaching hospitals; the Beaumont Sainsbury Animal Hospital in central London, the Queen Mother Hospital for Animals (Europe's largest small animal referral centre), the Equine Referral Hospital, and the Farm Animal Clinical Centre located at the Hertfordshire campus.
RVC Press Release 23 June 2017
See other Press Releases.
